How to open converted files?
Parquet files created by arcMS can be easily opened with
R or Python. There are also functions included in the arcMS
package to recreate sample_dataset objects from Parquet
files. This class of object is associated with generic functions to
quickly get data, metadata and simple plots like TIC.
1. Load converted Parquet files in R without arcMS
Files created by arcMS in the Parquet format can be
opened in R with the Arrow library.
Data can be loaded in RAM with the following commands:
library(arrow)
data = read_parquet("converted_file.parquet")
head(data)
#> # A tibble: 6 × 7
#> rt scanid mslevel mz intensity bin dt
#> <dbl> <int> <fct> <dbl> <dbl> <int> <dbl>
#> 1 0.00740 1 1 556. 0 19 1.35
#> 2 0.00740 1 1 556. 5 19 1.35
#> 3 0.00740 1 1 556. 7 19 1.35
#> 4 0.00740 1 1 556. 0 19 1.35
#> 5 0.00740 1 1 556. 0 23 1.63
#> 6 0.00740 1 1 556. 1 23 1.63Data can then be filtered and quickly aggregated, e.g. to obtain TIC plot:
library(data.table)
data = as.data.table(data)
datalow = data[mslevel == 1, ]
TIC = datalow[, list(intensity = sum(intensity)), by=list(rt)]
plot(TIC$rt, TIC$intensity, type = "l")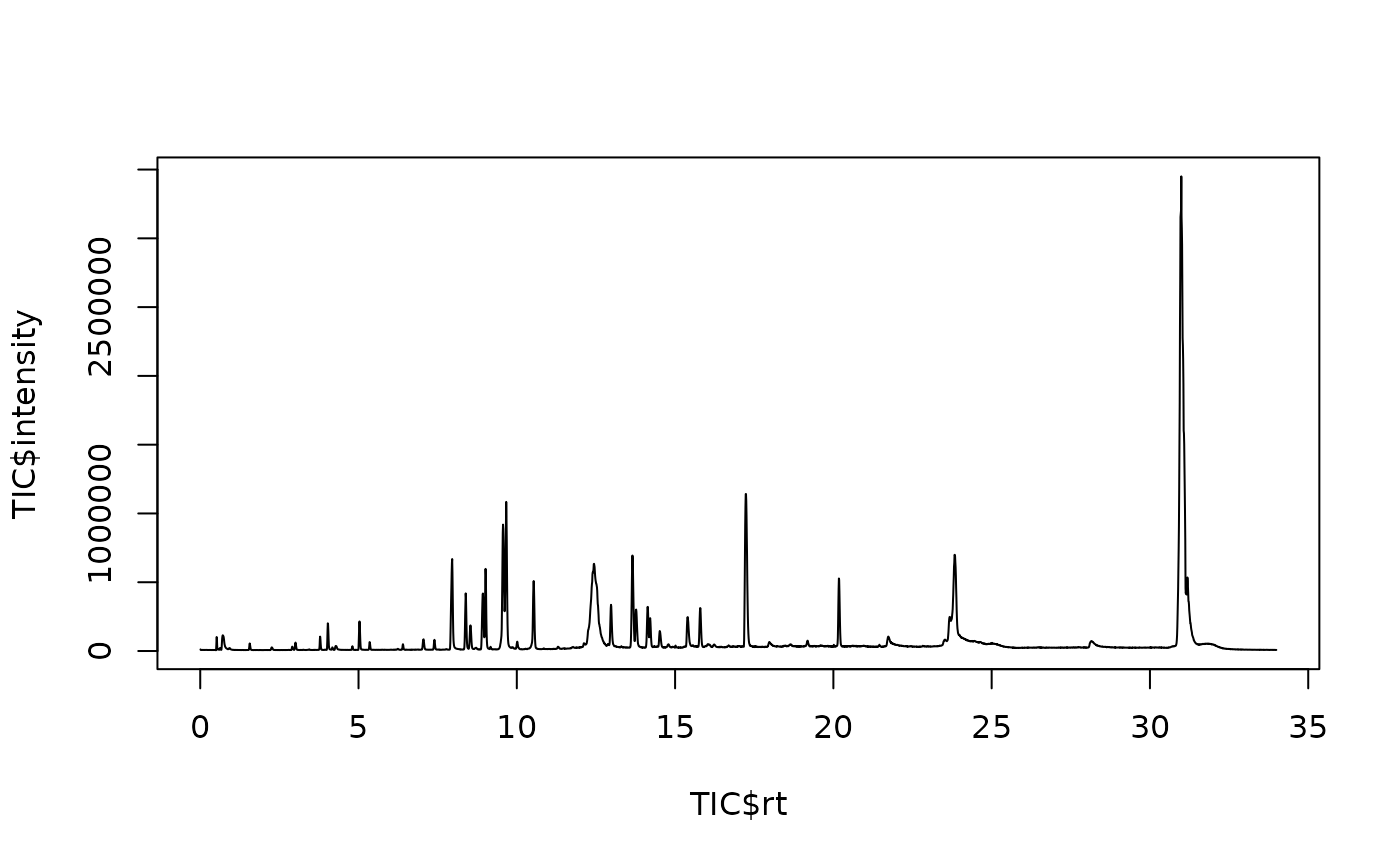
To save RAM, data can also be manipulated directly on-disk thanks to
the Arrow library (open_dataset() function):
data = open_dataset("converted_file.parquet")Data can be filtered, rearranged, sorted and aggregated with the
dplyr syntax, and only the resulting data will be loaded in RAM (with
the collect() function):
2. Load converted Parquet files in Python
Parquet files can be opened as a DataFrame in Python with the pandas library:
import pandas as pd
data = pd.read_parquet("converted_file.parquet")
ms1 = data[data['mslevel'] == "1"]
print(ms1)It can also be loaded as an Arrow object (ParquetDataset) with the pyarrow library:
3. Load converted Parquet files in R with arcMS
The methods above only retrieve the main data from the Parquet file, but not metadata. Other functions must be used to retrieve metadata.
To simplify opening both data and metadata, functions are available
in arcMS to load a Parquet file as a
sample_dataset object, also allowing easy manipulation with
some generic functions.
library(arcMS)
dataset = create_sample_dataset("converted_file.parquet") Retrieve main data:
data = get_sample_data(dataset)
head(data)
#> # A tibble: 6 × 7
#> rt scanid mslevel mz intensity bin dt
#> <dbl> <int> <fct> <dbl> <dbl> <int> <dbl>
#> 1 0.00740 1 1 556. 0 19 1.35
#> 2 0.00740 1 1 556. 5 19 1.35
#> 3 0.00740 1 1 556. 7 19 1.35
#> 4 0.00740 1 1 556. 0 19 1.35
#> 5 0.00740 1 1 556. 0 23 1.63
#> 6 0.00740 1 1 556. 1 23 1.63Retrieve sample metadata:
sample_metadata = get_sample_metadata(dataset)
str(sample_metadata)
#> Classes 'data.table' and 'data.frame': 1 obs. of 45 variables:
#> $ id : chr "0134efbf-c75a-411b-842a-4f35e2b76347"
#> $ name : chr "cal 1"
#> $ description : chr "NaN"
#> $ sample.description : chr "NaN"
#> $ sample.originalSampleId : chr "NaN"
#> $ sample.id : chr "8c6f3993-a791-4396-a299-b4dee758a5e7"
#> $ sample.gender : chr "NaN"
#> $ sample.name : chr "cal 1"
#> $ sample.assayConditions : chr "NaN"
#> $ sample.bracketGroup : chr "NaN"
#> $ sample.dose : chr "NaN"
#> $ sample.dosingRoute : chr "NaN"
#> $ sample.day : chr "NaN"
#> $ sample.eCordId : chr "NaN"
#> $ sample.experimentalConcentration : chr "NaN"
#> $ sample.groupId : chr "NaN"
#> $ sample.injectionId : chr "NaN"
#> $ sample.matrix : chr "NaN"
#> $ sample.solventDelay : chr "NaN"
#> $ sample.species : chr "NaN"
#> $ sample.studyId : chr "NaN"
#> $ sample.subjectId : chr "NaN"
#> $ sample.molForm : chr "NaN"
#> $ sample.preparation : chr "NaN"
#> $ sample.sampleType : chr "Standard"
#> $ sample.batchId : chr "NaN"
#> $ sample.studyName : chr "NaN"
#> $ sample.sampleLevel : chr "Unspecified"
#> $ sample.sampleWeight : num 1
#> $ sample.dilution : num 1
#> $ sample.replicateNumber : int 1
#> $ sample.wellPosition : chr "1:A,7"
#> $ sample.injectionVolume : num 10
#> $ sample.acquisitionRunTime : num 34
#> $ sample.acquisitionStartTime : chr "2021-11-17T16:14:36.1683419+01:00"
#> $ sample.time : chr "NaN"
#> $ sample.processingOptions : chr "QuantitationStd"
#> $ sample.processingFunction : chr "NaN"
#> $ sample.processingSequenceNumber : int 0
#> $ components.odata.navigationLink : chr "http://localhost:50034/unifi/v1/sampleresults(0134efbf-c75a-411b-842a-4f35e2b76347)/components"
#> $ spectra.odata.navigationLink : chr "http://localhost:50034/unifi/v1/sampleresults(0134efbf-c75a-411b-842a-4f35e2b76347)/spectra/mass.mse"
#> $ spectrumInfos.odata.navigationLink : chr "http://localhost:50034/unifi/v1/sampleresults(0134efbf-c75a-411b-842a-4f35e2b76347)/spectrumInfos"
#> $ chromatogramInfos.odata.navigationLink: chr "http://localhost:50034/unifi/v1/sampleresults(0134efbf-c75a-411b-842a-4f35e2b76347)/chromatogramInfos"
#> $ sampleName : chr "cal 1_replicate_1"
#> $ analysisName : chr "20211117_NORMAN_2e EIL_semi-quanti_Parent-TP products-testsAPI"
#> - attr(*, ".internal.selfref")=<externalptr>Retrieve spectrum metadata:
spectrum_metadata = get_spectrum_metadata(dataset)
str(spectrum_metadata)
#> Classes 'data.table' and 'data.frame': 3 obs. of 32 variables:
#> $ id : chr "a8d5230c-aaa3-4f34-9a20-738ae8484612" "896256ff-aa7e-4e45-b76c-5f4fb6275fd3" "b83dffcc-5817-4ff6-b535-f2ec0869e5e3"
#> $ name : chr "1: MS LockSpray Reference Data (546.2766-566.2766) 6V ESI+" "2: HD TOF MSe (50-1000) 6V ESI+" "3: HD TOF MSe (50-1000) 20-56V ESI+"
#> $ isCentroidData : logi FALSE FALSE FALSE
#> $ isRetentionData : logi TRUE TRUE TRUE
#> $ isIonMobilityData : logi FALSE TRUE TRUE
#> $ hasCCSCalibration : logi FALSE TRUE TRUE
#> $ detectorType : chr "MS" "MS" "MS"
#> $ analyticalTechnique.X.odata.type : chr "#Waters.WebApi.Common.Models.MSTechnique" "#Waters.WebApi.Common.Models.MSTechnique" "#Waters.WebApi.Common.Models.MSTechnique"
#> $ analyticalTechnique.hardwareName : chr "NaN" "NaN" "NaN"
#> $ analyticalTechnique.scanningMethod : chr "MS" "MS" "MS"
#> $ analyticalTechnique.massAnalyser : chr "QTOF" "QTOF" "QTOF"
#> $ analyticalTechnique.ionisationMode : chr "+" "+" "+"
#> $ analyticalTechnique.ionisationType : chr "ESI" "ESI" "ESI"
#> $ analyticalTechnique.lowMass : num 546 50 50
#> $ analyticalTechnique.highMass : num 566 1000 1000
#> $ analyticalTechnique.adcGroup.acquisitionMode : chr "ADC_PD" "ADC_PD" "ADC_PD"
#> $ analyticalTechnique.adcGroup.acquisitionFrequency : chr "NaN" "NaN" "NaN"
#> $ analyticalTechnique.tofGroup.nominalResolution : num 30000 30000 30000
#> $ analyticalTechnique.tofGroup.mseLevel : chr "Unknown" "Low" "High"
#> $ analyticalTechnique.tofGroup.pusherFrequency : num 14085 14085 14085
#> $ analyticalTechnique.quadGroup : chr "NaN" "NaN" "NaN"
#> $ axisX.label : chr "Observed mass" "Observed mass" "Observed mass"
#> $ axisX.unit : chr "m/z" "m/z" "m/z"
#> $ axisX.lowerBound : num 546 50 50
#> $ axisX.upperBound : num 566 1000 1000
#> $ axisY.label : chr "Intensity" "Intensity" "Intensity"
#> $ axisY.unit : chr "Counts" "Counts" "Counts"
#> $ axisY.lowerBound : chr "NaN" "NaN" "NaN"
#> $ axisY.upperBound : chr "NaN" "NaN" "NaN"
#> $ analyticalTechnique.adcGroup.ionResponses.ionType : chr "PEPTIDE" "PEPTIDE" "PEPTIDE"
#> $ analyticalTechnique.adcGroup.ionResponses.charge : int 1 1 1
#> $ analyticalTechnique.adcGroup.ionResponses.averageIonArea: num 5.92 5.92 5.92
#> - attr(*, ".internal.selfref")=<externalptr>